INSTITUT DE BIOLOGIE DE L'ÉCOLE NORMALE SUPÉRIEURE
Nous offrons des services de génomique en séquençage à haut débit. Nous sommes spécialisés sur les applications de génomique fonctionnelle, plus particulièrement l’analyse du transcriptome (RNA-seq) et du régulome (épitranscriptomique), chez tous les eucaryotes. Nous avons une expertise dans le séquençage en lecture longue (Nanopore).
Nous proposons aussi un service de séquençage « ready-to-load », pour les utilisateurs fabriquant leurs propres banques.

Nous proposons un service de RNA-seq avec séquençage lectures courtes Illumina (NextSeq 2000) et lectures longues (Oxford Nanopore Technologies MinION et PromethION).
Applications générales : Les données RNA-seq peuvent servir à quantifier les ARN, mesurer des différences d’expression de gènes ou isoformes, analyser des variants d’épissage ou des polymorphismes, assembler des transcriptomes de novo (si la séquence génomique n’est pas disponible ou l’annotation fragmentaire).
Nature des échantillons : Nous travaillons sur la plateforme avec de nombreuses espèces animales eucaryotes que ce soient des organismes modèles ou non (humain, rat, souris, poissons, grenouilles, mouches, papillons, meduses, levures, plantes…). Les échantillons sont les ARN totaux (nous ne proposons pas l’étape d’extraction des ARN), qui peuvent provenir de cellules (y compris triées), tissus (y compris fixés), biopsies…
Services proposés :
Nous proposons un service de scRNA-seq (10x Genomics) avec séquençage lectures courtes Illumina (NextSeq 2000) ou lectures longues (Oxford Nanopore Technologies MinION et PromethION) qui permet d’étudier les isoformes en cellule unique.
Applications générales : Les données scRNA-seq peuvent servir à explorer l’hétérogénéité des cellules au sein d’un tissu (y compris tumeurs), à détecter des sous-populations ou des types cellulaires rares.
Nature des échantillons : Nous travaillons sur la plateforme avec de nombreuses espèces animales eucaryotes que ce soient des organismes modèles ou non (humain, rat, souris, poissons, grenouilles, mouches, papillons, meduses, levures, plantes…). Les échantillons sont soit des cellules fraîches dissociées et/ou triées (seulement en interne à l’IBENS), ou des cellules fixées (nous participons à la mise au point de l’approche de fixation avec le protocole ACME).
Services proposés :
Nous proposons un service de fabrication de banques et la détection de bases modifiées sur l’ARN (m6A), par séquençage d’ARN direct (Nanopore).
Applications générales m6A : L’épitranscriptomique permet d’identifier et de quantifier des nucléotides modifiés au sein des ARN. La bases modifiée m6A (N6-methyladenine) est la modification la plus commune sur les ARNm eucaryotes.
Nature des échantillons : Nous travaillons sur la plateforme avec de nombreuses espèces animales eucaryotes que ce soient des organismes modèles ou non (humain, rat, souris, poissons, grenouilles, mouches, papillons, meduses, levures, plantes…). m6A : Les échantillons sont les ARN totaux (nous ne proposons pas l’étape d’extraction des ARN), qui peuvent provenir de cellules (y compris triées), tissus (y compris fixés), biopsies…
Services proposés :
Notre plateforme propose les protcoles de fabrication de banque suivants :
| Banques pour séquençage lectures courtes (Illumina) |
Banques pour séquençage lectures longues (Oxford Nanopore Technologies) |
|
|---|---|---|
| RNA-seq | Purification des ARNm polyA Déplétion des ARN ribosomiques (long ARNs non-codants) |
Purification des ARNm polyA |
| RNA-seq petites quantités | Amplification des échantillons de faible quantité | Amplification des échantillons de faible quantité |
| 10x Genomics 3’ Gene Expression | 10x Genomics 3’ Gene Expression (protocole scNauMi-seq) |
|
| Régulome |
N/A |
ARN direct (détection m6A) |
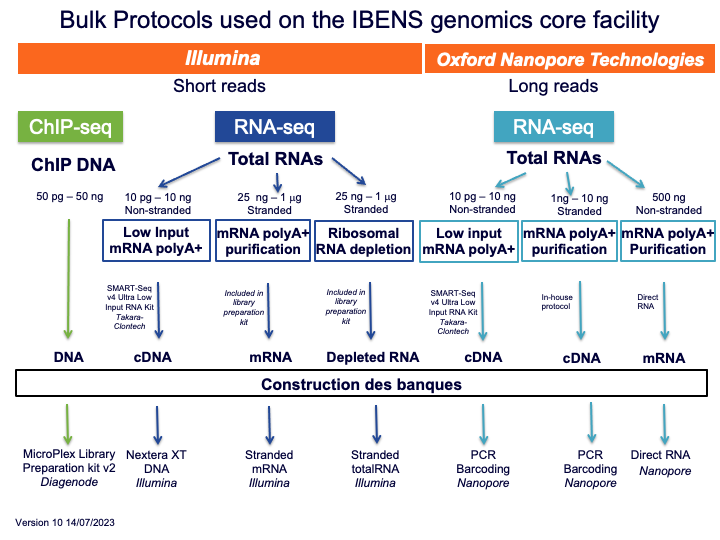
Le schéma suivant permet de déterminer le type de protocole "bulk" proposé par notre plateforme le plus adapté aux différentes problématiques :
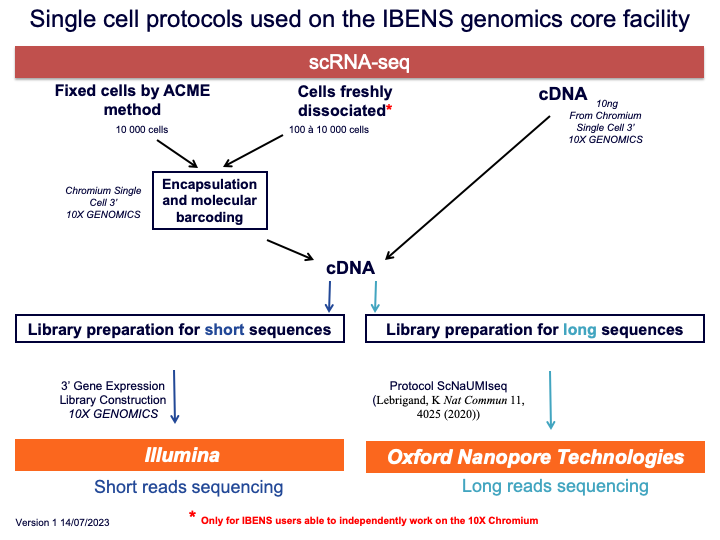
Voici l'ensemble des protocoles single-cell lectures courtes et longues proposés par notre plateforme :
Notre plateforme dispose de séquenceurs de deux fournisseurs différents : Illumina pour les lectures courtes et Oxford Nanopore Technologies pour les lectures longues.
Nous séquençons aussi les banques réalisées par nos utilisateurs (« ready-to-load »).
Les séquenceurs Illumina permettent de réaliser des séquençages appariés ou non. Lors d’un séquençage apparié, les deux extrémités des inserts entre les adaptateurs sont séquencées contre une seule lors d’un séquençage standard. Dans la plupart des cas, les longueurs des lectures appariées sont identiques (elles sont habituellement notées sous la forme 2 × 100).
La plateforme GenomiqueENS utilise actuellement pour ses services un séquenceur Illumina NextSeq 2000.
| Illumina NextSeq 2000 | |||||||
|---|---|---|---|---|---|---|---|
| Flowcell P1 (≃ 100 millions de lectures par run) |
Flowcell P2 (≃ 400 millions de lectures par run) |
Flowcell P3 (≃ 1,2 milliards de lectures par run) |
Flowcell P4 (≃ 1,8 milliards de lectures par run) |
||||
| Kit | Taille (pb) | Kit | Taille (pb) | Kit | Taille (pb) | Kit | Taille (pb) |
| N/A | N/A | N/A | 50 cycles | 1 × 50 2 × 25 |
|||
| 100 cycles | 1 × 100 2 × 50 |
100 cycles | 1 × 100 2 × 50 |
100 cycles | 1 × 100 2 × 50 |
100 cycles | 1 × 100 2 × 50 |
| N/A | 200 cycles | 2 × 100 | 200 cycles | 2 × 100 | 200 cycles | 2 × 100 | |
| 300 cycles | 2 × 150 | 300 cycles | 2 × 150 | 300 cycles | 2 × 150 | 300 cycles | 2 × 150 |
| 600 cycles | 2 × 300 | 600 cycles (*) | 2 × 300 | N/A | N/A | ||
Les séquenceurs d’Oxford Nanopore Technologies permettent de réaliser des séquençages d’ADN or d’ARN. Le nombre et la taille des lectures varient selon le type de séqueçages effectué :
| Oxford Nanopore Technologies MinION et PromethION P2 | ||||
|---|---|---|---|---|
| Kit | Taille (pb) | Nombre de lectures MinION |
Nombre de lectures PromethION |
|
| ADN (ADNc) |
Jusqu’à plusieurs centaines de kb (transcrits pleine longueur pour le RNA-Seq) |
6–8 millions de lectures | 80–120 millions de lectures | |
| ARN direct | 1 000 pb en moyenne | ≃ 1 millions de lectures | En cours de test | |
Notre plateforme propose deux niveaux d’accompagnement de ses utilisateurs au niveau bioinformatique (le détail des services d’analyses bioinformatiques est disponible dans les sections décrivant les différentes applications proposées) :
Les analyses bioinformatiques sont seulement réalisées sur les données produites par notre plateforme. Pour analyser des données séquencées ailleurs, nous vous conseillons de vous orienter vers une plateforme bioinformatique.
Depuis 2006, nous avons créé un système de « projet accompagné » sous forme de collaboration scientifique qui prend en charge tout le processus expérimental et vous fourni des résultats « prêts à publier ». Vous bénéficiez ainsi de notre expérience et de notre savoir faire depuis le dessin expérimental de vos expériences jusqu’à l’analyse de vos données et la participation à la rédaction des publications.
Nous sommes ouvert aux laboratoires publics comme aux entreprises privées. La majorité de nos projets sont effectués pour des personnes extérieures à l’IBENS.
La collaboration de recherche mise en place implique que le binôme expérimentation/analyse associé au projet figure dans la liste des auteurs de la première publication présentant les résultats obtenus lorsque la plateforme a participé à la fabrication des banques et/ou à l’analyse des résultats (sauf analyses automatisées). Ceci ne concerne pas les projets « ready to load ». L’affiliation à utiliser est la suivante :
GenomiqueENS, Institut de Biologie de l'ENS (IBENS), Département de biologie, École normale supérieure, CNRS, INSERM, Université PSL, 75005 Paris, France
Pour tous les projets (y compris les projets « ready to load »), la mention de la participation du réseau « France Génomique » devra apparaître dans la partie remerciements de la publication avec la phrase suivante :
The GenomiqueENS core facility was supported by the France Génomique national infrastructure, funded as part of the "Investissements d'Avenir" program managed by the Agence Nationale de la Recherche (contract ANR-10-INBS-09)