INSTITUT DE BIOLOGIE DE L'ÉCOLE NORMALE SUPÉRIEURE
We offer services in genomics and especially in high-throughput sequencing. We are specialized in functional genomics applications in all eukaryotes, particularly in Transcriptomics: RNA-seq (low input, long-read, single-cell) and Epigenomics/Epitranscriptomics: RNA m6A methylation. Our expertise also lies in long-read sequencing (Nanopore).
We also propose a sequencing service “ready-to-load” for users who make their own libraries.
| Applications | Operating |

We propose a RNA-seq service with short-read Illumina sequencing (NextSeq 2000) and long-read sequencing (Oxford Nanopore Technologies MinION and PromethION).
General applications: RNA-seq data can serve to quantify RNA, measure differences in gene expression or isoforms, analyse splicing variants or polymorphisms, assemble de novo transcriptomes (if the genomic sequence is not available or with fragmentary annotation).
Samples: We work with many eukaryotic animal species, whether they are model organisms or not (human, rat, mouse, fish, frog, fly, butterfly, jellyfish, yeast, plants…). Samples are total RNA (we do not perform the step of RNA extraction), which can be obtained from cells (included sorted cells), tissues (included fixed), biopsies…
Proposed services:
We propose a scRNA-seq service (10x Genomics) with short read Illumina sequencing (NextSeq 2000) or long read sequencing (Oxford Nanopore Technologies MinION and PromethION) enabling to study isoforms at the single cell level.
General applications: scRNA-seq data can serve to explore the heterogeneity of cells within a tissue (including tumors), detect sub-populations or rare cell types.
Samples: We work with many eukaryotic animal species, whether they are model organisms or not (human, rat, mouse, fish, frog, fly, butterfly, jellyfish, yeast, plants…). Samples are either fresh cells dissociated or sorted (only within IBENS), or fixed cells (we are involved in the development of an approach using the ACME protocol).
Proposed services:
We propose a service to construct the libraries for detection of modified nucleotides on RNA (m6A) with RNA direct sequencing (Nanopore).
General applications m6A: Epitranscriptomics enables to identify and quantify modified nucleotides within RNA. The modified base m6A (N6-methyladenine) is the most common modification in eukaryotic mRNA.
Samples: We work with many eukaryotic animal species, whether they are model organisms or not (human, rat, mouse, fish, frog, fly, butterfly, jellyfish, yeast, plants…). m6A: Samples are total RNA (we do not perform the step of RNA extraction), which can be obtained from cells (included sorted cells), tissues (included fixed), biopsies…
Proposed services:
Our genomcis core facility offers the following library preparation protocols:
| Short read library protocols (Illumina) |
Long read library protocols (Oxford Nanopore Technologies) |
|
|---|---|---|
| RNA-seq | PolyA mRNA purification rRNA depletion (long non-coding RNAs) |
PolyA mRNA purification |
| RNA-seq low input | Low input amplification | Low input amplification |
| 10x Genomics 3’ Gene Expression | 10x Genomics 3’ Gene Expression (scNauMi-seq protocol) |
|
| Regulome |
N/A |
Direct RNA (m6A modification detection) |
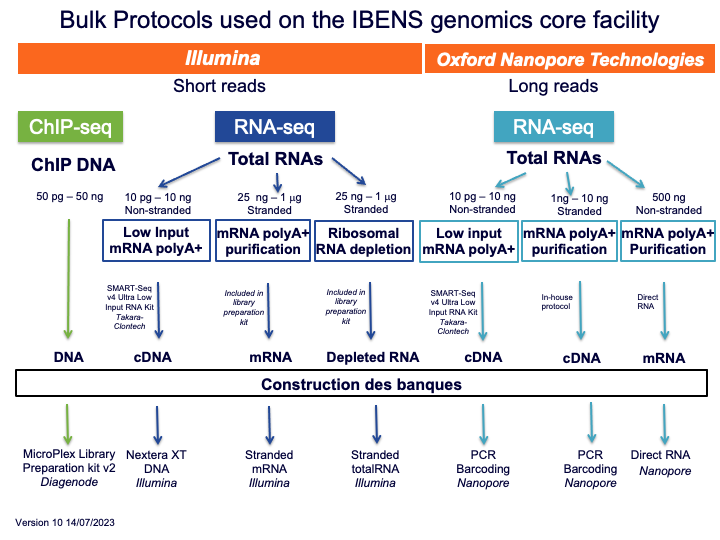
The following schema allow to choose the protocol against the biological question:
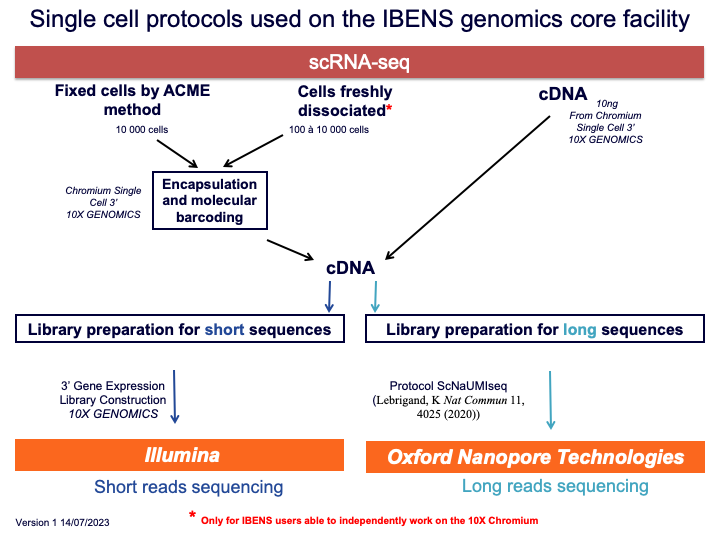
Here all the single-cell protocols available on our genomics core facility:
Our genomics core facility have sequencers from two supplier: Illumina for short reads sequencing and Oxford Nanopore Technologies for long reads sequencing.
We also propose a sequencing service “ready-to-load” for users who make their own libraries.
Illumina sequencers allow to perform single and or paired-end sequencing. With paired-end sequencing, the two ends of the inserts between adapters are sequenced instead of just one in single end mode. In most the the cases, the length of the two ends are the sames (they are commonly noted like 2 × 100).
The GenomiqueENS core facility currenly use an Illumina NextSeq 2000 sequencer for short reads sequencing.
| Illumina NextSeq 2000 | |||||||
|---|---|---|---|---|---|---|---|
| Flowcell P1 (~100 million reads per run) |
Flowcell P2 (~400 million reads per run) |
Flowcell P3 (~1.2 billion reads per run) |
Flowcell P4 (~1.8 billion reads per run) |
||||
| Kit | Size (bp) | Kit | Size (bp) | Kit | Size (bp) | Kit | Size (bp) |
| N/A | N/A | N/A | 50 cycles | 1 × 50 2 × 25 |
|||
| 100 cycles | 1 × 100 | 100 cycles | 1 × 100 2 × 50 |
100 cycles | 1 × 100 2 × 50 |
100 cycles | 1 × 100 2 × 50 |
| N/A | 200 cycles | 2 × 100 | 200 cycles | 2 × 100 | 200 cycles | 2 × 100 | |
| 300 cycles | 2 × 150 | 300 cycles | 2 × 150 | 300 cycles | 2 × 150 | 300 cycles | 2 × 150 |
| 600 cycles | 2 × 300 (*) | 600 cycles (*) | 2 × 300 | N/A | N/A | ||
Oxford Nanopore Technologies sequencers allow to sequence DNA or RNA. The read lengths and the throughput vary against the sequencing mode:
| Oxford Nanopore Technologies MinION and PromethION P2 | |||
|---|---|---|---|
| Kit | Size (bp) | MinION read count |
PromethION read count |
| DNA (cDNA) |
Until several thousands kb (full length transcripts in RNA-seq) |
6–8 million reads | 80–120 million reads |
| Direct RNA | ~1,000 bp mean size | ~1 million reads | Under testing |
Our genomics core facility offers two service levels for bioinformatics analysis (the descriptions of the bioinformatics services are available in the application sections of this page):
Bioinformatics analyses are only performed on data sequenced by our genomics core facility. To analyse your data, you can contact a bioinformatics core facility.
Since 2006, we have created an “accompanied project” service as a scientific collaboration that handle all the experimental process and provide “read-to-publish results”. You benefit from our experience and our know-how from experimental design to the data analysis and an help to publication writing.
We are open to academic laboratories and companies. Most of our project are performed with teams outside the IBENS.
Research collaboration implies that the two persons working on experiments and bioinformatics associated to the project are listed as co-authors the first time the results are published. This is only the case if the core facility has been involved in library preparation and/or bioinformatics analysis (except automated analyses), not in the case of “ready-to-load” projects. The affiliation to use is the following:
GenomiqueENS, Institut de Biologie de l'ENS (IBENS), Département de biologie, École normale supérieure, CNRS, INSERM, Université PSL, 75005 Paris, France
For all projects (including “ready-to-load” projects), reference to the "France Genomique” infrastructure must appear in the acknowledgements section of the article, with the following sentence:
The GenomiqueENS core facility was supported by the France Génomique national infrastructure, funded as part of the "Investissements d'Avenir" program managed by the Agence Nationale de la Recherche (contract ANR-10-INBS-09)